Pıhtılaşma (Hemostaz)
Pıhtılaşma (Hemostaz)
Hemostaz kanın pıhtışalması demektir. Damar cidarında herhangi bir hasar meydana geldiğinde birbiri ardısıra 3 olay meydana gelir:- Damarın büzüşmesi (damar fazı)
- Trombosit tıkacı oluşması (primer pıhtılaşma mekanizması-trombosit fazı)
- Fibrin trombüsü oluşması (sekonder pıhtılaşma mekanizması-plazma fazı)
Primer hemoztatik mekanizma

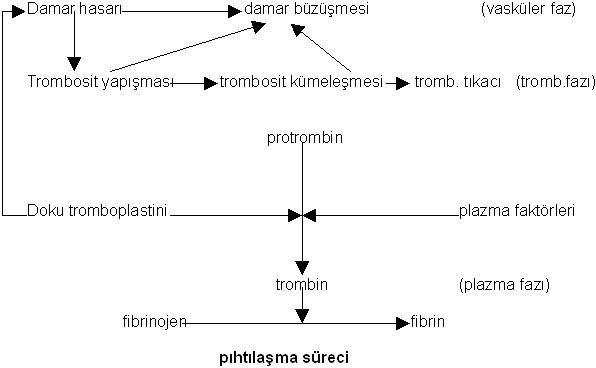
Sekonder hemostatik mekanizma
Bu fazda protrombinin trombine dönmesi sonucu geçici trombosit tıkacının kalıcı sağlam pıhtıya dönüşmesi gerçekleşir. Protrombinin trombine dönmesi iç ve dış yol denilen iki yol aracılığıyla meydana gelir. Her iki yol birbiriyle ilişkilidir ve ortak bir yolda birleşerek pıhtı oluşumunu sağlarlar.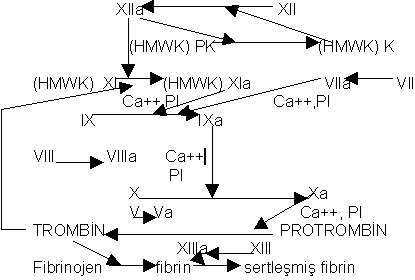 HMWK: yüksek molekül ağırlıklı kininojen PK: prekallikrein
HMWK: yüksek molekül ağırlıklı kininojen PK: prekallikreinFaktör VII'in doku faktörüne bağlanarak Ca++ yardımıyla başlattığı pıhtılaşma yolu dış yolu, PK/HMWK-Kinonojenin F XII'yi aktive etmesiyle başlayan yol ise iç pıhtılaşma yolunu oluşturur.
Laboratuar testlerinden PT dış yol aracılığıyla trombin oluşumunu, PTT testi ise iç yoldan trombin oluşumunu ölçen testlerdir.
Pıhtılaşmanın mekanizmaları arasındaki ilişki
Hemostazın üç fazı birbirinden bağımsız değil birlikte çalışırlar. Pıhtılaşmanın gerçekleşmesini sağlayan faktörler olduğu kadar aşırıya kaçmasını engelleyen faktörler de bir denge içinde görev yaparlar. Örneğin endotel hücreleri prostoglandin I2, nitrik oksit ve ADP adlı maddeleri salgılayarak trombositlerin biraraya gelmesini engellerlerken kollagen, fibronektin ve von Willebrand faktörünü salgılayarak pıhtılaşmayı başlatırlar. Ortaya çıkan fibrin tıkacı yine damar endotel hücrelerinden salgılanan doku plazminojen aktivatörü ile çözünmeye başlar ve aynı zamanda heparin ve trombomodülin de salgılanarak trombinin oluşumu engellenir. Bir başka engelleyici madde olan doku plazminojen aktivatör inhibitörü ise fibrinin çözülmesini engeller.Trombositler pıhtılaşmada temel görevlerden birini görürler. Trombositler sadece yaralanan yerde tıkaç oluşturmakla kalmazlar, içlerindeki granüllerden salgıladıkları maddelerle hem damar büzüşmesine katkıda bulunur, hem diğer trombositlerin olay yerine gelmesini sağlar, hem iç pıhtılaşma yolunun başlaması için gerekli maddeleri ortama salar hem de pıhtılaşma faktörlerinin aktiflenmesi için gerekli fosfolipid yüzeyi oluştururlar. İç ve dış yolların birleşip ortak yolu oluşturmasından sonra meydana gelen trombin geriye dönerek trombositlerin bir araya gelmesine katkıda bulunurken aynı zamanda daha fazla trombin oluşmasına da yardımcı olur.
Fibrinin eritilmesi (fibrinoliz)

TA inh: doku aktivatörü engelleyicisi
EACE: epsilon amino kaproik asit
ATIII:antitrombin III
İki uçlu ok: engelleyici etki
Yenidoğanda pıhtılaşma
Yenidoğanda pıhtılaşma sistemi daha büyük çocuklara ve erişkinlere göre daha az gelişmiştir. Örneğin:- Kan damarları daha kırılgandır
- Endotel hücrelerinden daha fazla prostasiklin yapılır
- Tormbosit yapışması von Willebrand faktörünün ve HMWK'nin daha fazla yapılması nedeniyle daha fazladır
- Trombositlerin biraraya gelmesinde bozukluk vardır:
- Epinefrine cevap azdır
- Ristosetine cevap fazladır
- Trombosit aktivasyonu fazladır
- Kanama zamanı normal sınırlar içindedir
- Trombin oluşumunda ve oluşumun engellenmesinde gecikmeler vardır, pıhtılaşma proteinlerinin kandan temizlenmesi de gecikmiştir
- Vitamin K ya bağlı pıhtılaşma faktörleri faktör XI ve XII, PK ve HMWK azalmıştır
- ATIII, Protein C (PC) ve Protein S (PS) azalmıştır
- Pıhtı erimesi ilgili maddelerin az yapılması nedeniyle bozuktur
- Pıhtı oluşumunu engelleyen PC, PS, ATIII ve heparin kofaktör II doğuştan eksiktir
Pıhtılaşma sorununun saptanması
Hastada pıhtılaşma sorunu olup olmadığının anlaşılması için- Detaylı öykü alınması
- Tam bir fizik inceleme yapılması
- Tam kan sayımı ve mümkünse parmak ucundan periferik yayma yapılması
- Pıhtılaşma testlerinin yapılıp yorumlanması gerekir
Ameliyat öncesi pıhtılaşma sisteminin değerlendirilmesi
- Öykü:- Negatif ise sadece tam kan sayımı yeterlidir, başka test yapılması gerekmez
- Pozitif ise veya güvenilir değilse tam kan sayımı, kanama zamanı, PT, aPTT ve fibrinojen testleri yapılmalıdır
- Anormal sonuçlanan testler daha ileri araştırmayı gerektirir.
Edinilmiş Kanama Bozuklukları
Vitamin K eksikliği
Normal miyadında yenidoğanda II, VII, IX ve X numaralı pıhtılaşma faktörleri erişkinlere göre daha düşük miktarlardadır. Yaşamın ilk günlerinde bu faktörlerin miktarı daha da düşerek 3. günde en düşük düzeylerine iner. Bunun nedeni doğumda vücutta depolanan K vitamini miktarının düşük olmasıdır. İnek sütündeki K vitamini anne sütüne göre 4 kat daha fazladır. Depoların düşük olması ve anne sütü ile beslenme eğer yeni doğana K vitamini takviyesi yapılmazsa hemorajik hastalık dediğimiz tablo ile sonuçlanır. Bu tablo yaşamın 2.-4. günleri arasında mide ve barsaktan kanama, göbekten kanama veya iç organlara kanama şeklinde bulgu verir. Hastalık K vitaminine cevap verdiği içöin tüm yenidoğanlara koruma amacıyla K vitamini verilmelidir.Annenin aldığı bazı pıhtılaşma önleyici ilaçlar veya epilepsi ilaçları da yenidoğanda F II, IX ve X miktarlarını azaltabilir. Kusma, ağır ishal, barsaktan emilim bozukluğu ile giden hastalıklar (Çöliyak hastalığı, kistik fibroz, safra kanalı atrezisi vb), akut ve kronik karaciğer hastalıkları ve uzun süre antibiyotik alımı da K vitamini eksikliği ve kanama meydana getirebilir.Karaciğer hastalığı
K vitamini yeterli düzeydeyken bile pıhtılaşma faktörlerinin yapımını engelleyen geçici bir karaciğer problemi bile yenidoğanda kanamalı hastalık meydana getirebilir ve bu durum K vitaminine cevap vermez. Karaciğerin enfeksiyonu, oksijensiz veya kansız kalması gibi durumlar bu olayı başlatabilir. Bu küçük prematüre bebeklerde daha sık görülen bir durumdur. Beklerde akciğer ve beyin gibi hayati organlara kanama meydana gelebilir ve ölüm riski yüksektir. Bu tabloda K vitaminine bağlı faktörler, F V ve fibrinojen azalmıştır. Hastalık sadece taze donmuş plazma ve kriyopresipitat ile tedavi edilebilir.Yaygın damar içi pıhtılaşması
Damar içinde trombositlerin ve pıhtılaşma faktörlerinin tüketilmesi durumudur. Damar içinde yaygın olarak fibrin tıkaçları ve kanama meydana gelir. Damar içinde alyuvarların da parçalanması mikroanjiopatik hemolitik anemi ile sonuçlanır. Pıhtılaşma sisteminin yaygın aktivasyonu tüm faktörlerin ve trombositlerin hızla tüketilmesine yol açar.- PT ve aPTT uzamış bulunur
- Fibrinojen miktarı azalmıştır
- Trombosit sayısı azalmıştır
- Periferik yaymada parçalanmış alyuvarlar görülür
- Fibrin yıkım ürünleri artmıştır
- F V, VIII ve XIII azalmıştır
Bu tabloya yol açabilecek durumlar arasında:
- Yanıklar
- Büyük cerrahi girişimler
- Travma
- Kanser
- Amniotik sıvı veya yağ embolisi
- Enfeksiyonlar (bakteriyel, viral, protozoal)
- Doğum sonrası böbrek yetmezliği
- Doğum kontrol hapı kullanımı
- Dev hemanjiom
- Siroz
- Uygunsuz kan nakli
- İlaç alerjisi sayılabilir.
Yaygın damariçi pıhtılaşmasının tedavisi için öncelikle neden olan hastalığın düzeltilmesi gereklidir. Ayrıca hastaya taze donmuş plazma, trombosit ve eritrosit suspansiyonu desteği verilmelidir.
Kalıtsal pıhtılaşma faktörü eksiklikleri
Hemofili A ve B
En sık görülen pıhtılaşma faktörü eksikliği hemofili A ve B dir. X kromozomuna bağlı resesiv kalıtımla geçerler. Hemofili A FVIII, hemofili B FIX eksikliğidir. Hemofilinin sıklığı 100.000 erkekte 20 olarak saptanmıştır. Olguların %80-85'ini hemofili A, geri kalanını hemofili B oluşturur. Hemofiliden sorumlu yüzlerce nokta mutasyonu ve gen inversiyonu vardır. kadınlar genellikle taşıyıcıdırlar ama bazı durumlarda hasta da olabilirler (diğer X kromozomunun inaktive olması, somatik mozaisizm). Hemofilinin klinikte karşımıza çıkış şekli A ve B tipinde birbirinin aynıdır. Faktör düzeyi < %1 olan hastalar ağır hemofili olarak kabul edilirler ve aile öykülerinin pozitif olması yanı sıra bu hastalarda klinik bulgular 12-18 aylıkken başlar. Orta derece hemofili olanlarda faktör düzeyleri %1-5 arasındadır. Bu hastalar ilk belirtilerini 2-5 yaş arasında yaşarlar. Hafif hemofili hastalarında ise faktör düzeyi %5'in üzerindedir ve bu hastalar ancak büyük bir travma veya cerrahi sırasında kanama ile klinik bulgu verirler. Tüm ağır hemofilihastalarında eklem içine kendiliğinden kanama görülebilir ancak sıklıkla bebek hareket etmeye başladıktan sonra yani 9 aydan sonra kanama görülür. Yenidoğan bebeklerde doğum sırasında kafa içine kanama görülebilir. Vakum veya forseps ile doğum risk oluşturur. Sezeryan kanama riskini ortadan kaldırmaz. Sünnet sonrası kanama hemofilide tipik bir klinik tablo olarak bilinmesine rağmen ağır hemofili hastalarının yarısından azında kanama görülür. Bu nedenle sünnet sonrası kanama olmaması hemofiliyi ekarte ettirmez. En sık eklem ve kas içine kanamalar görülür. Beyin, gastrointestinal sistem, idrar yolları ve karın içi kanamaları da görülebilir. En sık etkilenen eklemler diz, dirsek ve ayak bileği eklemleridir. Her iki eklemin de tutulması seyrek değildir. Eğer bir ekleme birden fazla kanama meydana gelirse o eklem hedef eklem olarak adlandırılır.. Kanama olduğunda eklem şiş ve ağrılıdır ve bükük pozisyonda tutulması tercih edilir. Eklem kanamalarına uygun şekilde yaklaşılmazsa eklemde kronik hemofilik artropati dediğimiz durum meydana gelir. Bunun daha da ileri aşaması ağrının da ortadan kalktığı eklem sertleşmesi tablosudur.Faktör VIII ve IX'un normal düzeyleri %50-%150 arasındadır. Normalin üzerinde düzeyler gebelikte, yaşlılıkta ve doğum kontrol hapı kullanımı sırasında görülür. sağlıklı normal veya prematüre bebeklerde faktör düzeyleri karaciğerin henüz olgunlaşmamış olması nedeniyle normalin %20-%50'si kadardır ve ancak 6. ayda erişkin düzeyine çıkar.Hastalıktan şüphelenildiğinde ilk yapılacak testler pıhtılaşma zamanı ve aPTT testidir. Bu testlerde uzama bulunması durumunda ikinci aşamada faktör düzeylerine bakılır. Ayırıcı tanıya von Willebrand hastalığı, trombosit işlev bozuklukları, edinilmiş hemofili ve F V, VII, XI ve fibrinojen eksiklikleri girer. Cerrahi, travma, diş çekimi sonrası kanama, eklem içine kanama hemofiliyi düşündürürken deri ve mukoza kanamaları von Willebrand hastalığını ve trombosit işlev bozukluklarını akla getirmelidir. Hastalığın ailenin daha çok erkek bireylerinde görülmesi hemofiliyi, kız erkek fark etmeden her nesilde ortaya çıkması ise von Willebrand hastalığını telkin eder.
Hemofilinin tedavisi eksik olan faktörün yerine konması ile gerçekleştirilir. Seçilecek ilaç ve doz eksikliğin derecesine, kanamanın yeri ve şiddetine göre değişir. Ufak kanamalarda faktör düzeyinin normalin %20-30'una çıkartılması yeterken ağır kanamalarda en az %50'ye çıkarmak gerekir. Cerrahi girişimler sırasında veya yaşamı tehdit eden kanamalarda faktör %75-100 düzeyine yükseltilmelidir. Yerine koyma tedavisi hastanın kilosuna göre hesaplanan bir doz verilerek yapılır. İnfüzyon aralıklı bolus şeklinde yapılabileceği gibi devamlı damar içi verilmesi şeklinde de uygulanabilmektedir. İkinci yol giderek daha fazla uygulanmakta olup böylelikte verilecek faktör miktarlarından da ekonomi yapılabilmektedir. Tedavi süresi ağrının ortadan kalkmasına ve eklem hareketlerinin düzelmesine bağlıdır. Tedavi yumuşak doku kanamalarında kan geri emilene kadar devam etmelidir. Büyük cerrahilerden sonra kanamanın kontrol edilmesi ve yara iyileşmesinin kolaylaştırılması amacıyla faktör verilmesi 10-14 gün devam ettirilmelidir. Faktör yerine bazen antidiüretik hormon olan desmopressin (DDAVP) kullanılır. İlaç faktör VIII'in aktivitesini artırmaktadır. Damar veya burun içine verilerek kullanılabilir. Etkisi 30-60 dakika sonra başlar. Hafif hemofililerde kanamayı durdurmak için kullanılabilir. 8-12 saatte bir tekrar etmek gerekebilir. Gastrointestinal veya diş kanamaları gibi yüzey kanamalarında faktöre ilave olarak antifibrinolitik ajanlar kullanılmalıdır. Epsilon amino kaproik asit ve traneksemik asit böyle ilaçlardır. Hemofili B de ancak çok yüksek saflıkta faktör ile birlikte verilebilirler, aksi taktirde damar içinde pıhtı oluşturabilirler. Eklem kanamalarında faktör verilmesine ek olarak kesin istirahat, eklemin yüksekte tutulması ve buz uygulaması gibi önlemler de alınmalıdır. Ağrı ve şişlik geçer geçmez eklem hareket ettirilmeli ve isometrik eksersizlere başlanmalıdır. Ağrı kesici olarak aspirin veya steroid olmayan antienflamatuar ilaçlar kullanılmamalıdır. En sık ölüme yol açan kanama kafa içine olan kanamadır. Şüphelenildiğinde hemen faktör infüzyonuna başlanmalı daha sonra görüntüleme yapılmalıdır. Faktör düzeyleri %100'e çıkartılmalı ve 14 gün kadar bu düzeyde tutulmalıdır.
Hastalığın doğum öncesi tanınması çok önemlidir, zira bu gerçekleştirilirse sadece gebeliğin sonlandırılması için karar verilebilmekle kalınmaz aynı zamanda bebeğe anne karnındayken faktör de verilebilir. Bebeğin kanından 16. haftadan önce faktör tayini yapılamaz ve annenin kanı ile bulaşma ve yanıltma riski taşır. En uygun yöntem 12. haftada korion villüs biyopsi mataryalinden gen analizi yaparak hastalığın geninin saptanmasıdır. Bunu yapmadan önce annenin faktör düzeylerinin ölçülmesi ve %40'ın altındaysa anneye faktör verildikten sonra yapılması unutulmamalıdır.
Hemofil A hastalarının yaklaşık %25'inde inhibitör gelişir. İnhibitör faktör VIII'e karşı gelişen antikorlardır. Tekrarlanan faktör infüzyonları sonrası ortaya çıkan bu durumun, eğer inhibitör düzeyi düşükse, yüksek doz faktör verilerek giderilmesi mümkündür. Eğer inhibitör yüksek titrelerdeyse bu tür hastalarda kanamanın durdurulması için devamlı yüksek doz faktör infüzyonu, protorombin ve aktive protrombin kompleks konsantreleri verilmesi, rekombinant faktör VIIa verilmesi ve plazmaferez gibi yöntemler uygulanmalıdır.
Von Willebrand Hastalığı
von Willebrand hastalığı (vWH) hasar görmüş damar cidarına trombositlerin yapışmasını sağlayan bir madde olan von Willebrand faktörünün (vWF) eksikliği nedeniyle ortaya çıkan bir hastalıktır. Bu hastalıkta cilt ve mukoza kanamaları, cerrahi sonrası veya travma sonrası kanamalar görülür. Hastalık ilgili faktörün doğuştan eksikliği ile meydana gelebileceği gibi işlevinin bozukluğu nedeniyle de ortaya çıkabilir. vWF damar endotel hücrelerinde ve megakaryositlerde yapılan ve endotel hücrlerinin ve trombositlerin içinde yer alan granüllerde depolanan bir glikoproteindir. vWH'nın çeşitli tipleri vardır.Tip 1, 2A ve 2B
Tip 1 en sık görülen tiptir ve bu hastalıkta vWF'ün hafif veya ortat derecede eksikliği söz konusudur. Faktör yapısal olarak normaldir. Ristosetin kofaktör (R:Co) ve F VIII düzeyleri de orantılı olarak azalmıştır.Tip 2A da plazmada ve trombositlerde yüksek molekül ağırlıklı vWF multimerleri azalmıştır ve bu nedenle trombositlerin damar endoteline yapışması bozulmuştur.Tip 2B daha nadir bir formudur ve yüksek molekül ağırlıklı multimerlerin kısmi kaybı ile karakterizedir. Bozukluk vWF'ün kendisindedir. Bir de yalancı vWH vardır ki bunda bozukluk faktörde değil trombositlerin yapışmayı sağlayan GP1b reseptöründedir. Bu hastalıkta vWF trombosit reseptörlerina fazla miktarda bağlanarak trombositleri dolaşımdan uzaklaştırır ve plazmada vWF miktarı düşerken trombosit agregasyonu artmış bulunur.
Tip 2N ve 2M
Tip 2N'de vWF'deki bir bozukluk sonucu FVIII'e bağlanmasında bir sorun vardır. bu nedenle bağlı olmayan FVIII dolaşımdan hızlıca uzaklaştırılır ve FVIII düzeyi düşer. Bu hastaların bir kısmana yanlışlıkla hemofili A tanısı konur. Desmopressin bu hastaların FVIII düeyini yanlızca geçici olarak yükseltir. Tedavide yüksek miktarda vWF içeren FVIII ürünleri kullanılır. Tip 2M vWH 'da ise vWF üzerindeki trombosit antijenine bağlanma yerinde bir bozukluk söz konusudur ve ristosetin kofaktör aktivitesi azalmıştır. Desmopressin ufak kanamaları durdurur ancak büyük travmalar veya cerrahi sırasında tip 2N deki gibi vWF den zengin FVIII kullanılmalıdır.Tip 3
Hemostazın primer ve sekonder kısmında bozuklukla karakterize ağır bir kanama hastalığıdır. Hem FVIII, hem de vWF düzeyleri saptanamayacak kadar düşüktür.Hemostazı engelleyen edinilmiş antikorlar
Hemofili A ve B hastalarında gelişen antikorların benzeri birçok başka klinik hastalıkta diğer pıhtılaşma faktörlerine karşı da gelişebilir.vWF'e karşı gelişen antikor
Wilms'tümörü, otoimmün hastalıklar (sistemik lupus eritematosus) veya lenfoproliferatif bozukluklarda ve anjiodisplastik lezyonlarda vWF'e karşı antikorlar gelişerek edinilmiş vWH meydana gelebilir. Altta yatan hastalığın düzeltilmesi vWF bozukluğunun da ortadan kalkması ile sonuçlanır.Lupus antikoagulan-antifosfolipid sendromu
İlk defa aPTT uzaması ile sitemik lupus eritematozus (SLE) hastalarında tarif edilen LA, faktör Xa ve Va, fosfolipid, kalsiyum'un fosfolipid bölgelerine bağlanarak fosfolipide bağlı pıhtılaşmanın vücut dışında uzamasına neden olur. Bu inhibitör aslında bir antikoagulan değildir. Bu nedenle LA bulunan hastalarda kanamaya yatkınlık oluşmaz. Hatta bazı hastalarda kanamanın aksine damar içinde pıhtı dahi oluşabilir. Bazen ITP'li hastalarda ortaya çıkan LA, HLA DR7 ve DRW35 antijeni pozitif olan kişilerde daha sık görülür. Lupus antikoagulanı aşağıdaki durumlarda ortaya çıkabilir.- HIV enfeksiyonu
- Araya giren viral enfeksiyonlar
- Sistemik antibiyotik tedavisi
- Diğer bazı ilaçlar (fenotiazin, hidralazin, prokrinamid, kinidin)
- SLE
- Lupus benzeri kronik otoimmün hastalıklar
- Belirgin nedeni olmayan arteriyel veya venöz trombozlar
- Tekrarlayan düşükleri olan kadınlar
LA sıklıkla IgG, daha az IgM veya IgA karakterinde bir immünglobülindir. LA varlığında aPTT testi uzamış bulunur ve normal plazma ile karışım testi yapıldığında aPTT normale dönmez.Cerrahi girişimlerden önce, özellikle de akut bir viral enfeksiyon da geçirilmişse, uzamış aPTT varlığında saptanan LA pozitifliği sonrası hastaya birçok başka laboratuar testi yapılması gereksizdir. Birkaç ay sonra tekrarlanan testte LA'nın kaybolduğu görülür.
Trombotik Hastalıklar
Damar sistemi içinde anormal bir pıhtı oluşmasına tromboz denir. Pıhtılaşma sisteminin fizyolojik işlevinde bir aşırıya kaçma ve kanama ve pıhtılaşma arasındaki dengenin bozulması sonucu tromboz meydana gelerek damar sisteminin tıkanmasına neden olabilir.Bir kişinin kendisinde veya aile öyküsünde aşağıdaki durumların olması tromboz oluşmasına yatkınlığa işaret eder.- Ailede tromboz öyküsü bulunması
- Tekrarlayan spontan düşükler
- Nadir vücut bölgelerinde tromboz oluşması
- Erken yaşta tromboz oluşması
- Antikoagulan tedaviye direnç
- Kumarinle nekroz meydana gelmesi
- Tekrarlayan spontan trombozlar
- Gebelik sırasında tromboz
- Göçeden yüzeyel tromboflebit
Venöz tromboz
Venöz tromboz kan akımının yavaş olduğu yerlerde ve bu akımın daha da yavaşladığı veya durduğu bölgelerde meydana gelir. Damar hasarı olsun veya olmasın pıhtılaşma sisteminin harakete geçmesi ile oluşur. Kan dolaşımında ciddi bir tıkanıklık meydana getirir. En önemli sonucu tromboembolidir. Yani bu pıhtıdan kopan parçaların başka organların damarlarını tıkamasıdır.Venöz tromboza yatkınlık oluşturan durumlar şunlardır:- Edinilmiş
- Travma
- Kan alınması
- İntravenöz kateterler
- Cerrahi veya kaza ile olan travmalar
- Isı hasarı (yanıklar)
- Enfeksiyonlar
- Ağır sıvı kaybı
- Şok
- Hareketsizlik
- Kanser
- L-asparaginaz tedavisi
- Doğum kontrol hapları
- Gebelik
- Syanotik kalp hastalığı
- Nefrotik sendrom
- Karaciğer hastalığı
- Diabetik anne çocuğu
- Sistemik lupus eritematosus
- Antikardiyolipin antikorları
- Lupus antikoagulan
- Kanı koyulaştıran hastalıklar
- Polisitemia vera
- Kronik myeloid lösemi
- Esansiyel trombositoz
- Paroksismal noktürnal hemoglobinüri
- Hiperlökositoz (akut lösemi)
- Orak hücre hastalığı
- K vitaminine bağımlı faktörlerin infüzyonu.
- Konjenital
- Antitrombin III eksikliği
- Faktör V Leiden
- Protein C eksikliği
- Protein S eksikliği
- Plazminojen eksikliği
- Disfibrinojenemi
- F XII eksikliği
- Prekallikrein eksikliği
- Heparin kofaktör II eksikliği
Tedavide yüzeyel tromboflebitlerde ağrı kesiciler, sıcak kompresler ve istirahat ile etkilenen uzvun yüksekte tutulması uygulanır.Derin ven trombozunda ise 7-10 gün tam doz heparin tedavisinden sonra uzun süre warfarin kullanılır.
Akciğer embolisinde tromboz eritici tedavi verilir. Tıbbi tedavi başarısız olursa cerrahi olarak pıhtını çıkartılması işlemi uygulanır.
Arteriyel tromboz
Sıklıkla damar cidarında bir hasar meydana gelmesiyle ve kan akımının hızlı olduğu yerlerde oluşur. En ciddi sonuç ise damarın tıkanmasıdır. Arteriyel pıhtının oluştuğu durumlar:- Edinilmiş
- Damar hasarı
- Travma
- Damar hastalığı
- Enfeksiyon
- Periarteritis nodosa
- Sistemik lupus eritematosus
- Şok
- Trombotik trombositopenik purpura
- Hemolitik üremik sendrom
- Orak hücre hastalığı, hemoglobin SC hastalığı
- Nefrotik sendrom
- Diabet
- Kan yağlarının artması
- Kalp hastalığı
- Kanı koyulaştıran hastalıklar (bak. Venöz tromboz)
- Konjenital/kalıtsal
- Homosistinüri
- Marfan sendromu
- Ailevi hiperkolesterolemi
- Mitral valve prolapsus
Tedavide cerrahi trombektomi embolaktomi uygulanır. Cerrahi yaklaşım mümkün değilse o zaman heparin ve pıhtı eritici ilaçlar kullanılır. Ek olarak trombosit işlevini engelleyen aspirin ve dipridamol gibi ilaçlar devreye sokulur.